



RT-qPCR розроблено на основі звичайної технології ПЛР.Він додає флуоресцентні хімічні речовини (флуоресцентні барвники або флуоресцентні зонди) до традиційної реакційної системи ПЛР і виявляє процес відпалу та розширення ПЛР у режимі реального часу відповідно до їх різних люмінесцентних механізмів.Зміни флуоресцентного сигналу в середовищі використовуються для розрахунку кількості зміни продукту в кожному циклі ПЛР.В даний час найпоширенішими методами є метод флуоресцентного барвника і метод зонда.
Метод флуоресцентного фарбування:
Деякі флуоресцентні барвники, такі як SYBR Green Ⅰ, PicoGreen, BEBO тощо, самі по собі не випромінюють світло, але випромінюють флуоресценцію після зв’язування з малою борозенкою дцДНК.Тому на початку ПЛР-реакції апарат не може виявити флуоресцентний сигнал.Коли реакція переходить до стадії відпалу-подовження (двоетапний метод) або стадії подовження (триетапний метод), у цей час подвійні ланцюги відкриваються, і нова ДНК-полімераза. Під час синтезу ланцюга флуоресцентні молекули об’єднуються в малій борозенці дцДНК і випромінюють флуоресценцію.У міру збільшення кількості циклів ПЛР все більше і більше барвників поєднується з дцДНК, і флуоресцентний сигнал також постійно посилюється.Візьміть SYBR Green Ⅰ як приклад.
Метод зонду:
Зонд Taqman є найбільш часто використовуваним зондом гідролізу.На 5'-кінці зонда є флуоресцентна група, зазвичай FAM.Сам зонд є послідовністю, комплементарною цільовому гену.На 3'-кінці флуорофора є група гасіння флуоресценції.Відповідно до принципу резонансної передачі енергії флуоресценції (Förster resonance energy transfer, FRET), коли репортерна флуоресцентна група (донорна флуоресцентна молекула) і гасить флуоресцентна група (акцепторна флуоресцентна молекула) Коли спектр збудження перекривається, а відстань дуже близька (7-10 нм), збудження донорної молекули може індукувати флуоресценцію молекули акцептора, при цьому автофлуоресценція послаблюється.Тому на початку ПЛР-реакції, коли зонд вільний і неушкоджений у системі, репортерна флуоресцентна група не буде випромінювати флуоресценцію.Під час відпалу праймер і зонд зв'язуються з шаблоном.На стадії подовження полімераза безперервно синтезує нові ланцюги.ДНК-полімераза має 5'-3' екзонуклеазну активність.Дійшовши до зонда, ДНК-полімераза гідролізує зонд із матриці, відокремить репортерну флуоресцентну групу від гасильної флуоресцентної групи та випустить флуоресцентний сигнал.Оскільки між зондом і шаблоном існує взаємозв’язок один-до-одного, метод зонда перевершує метод барвника з точки зору точності та чутливості тесту.
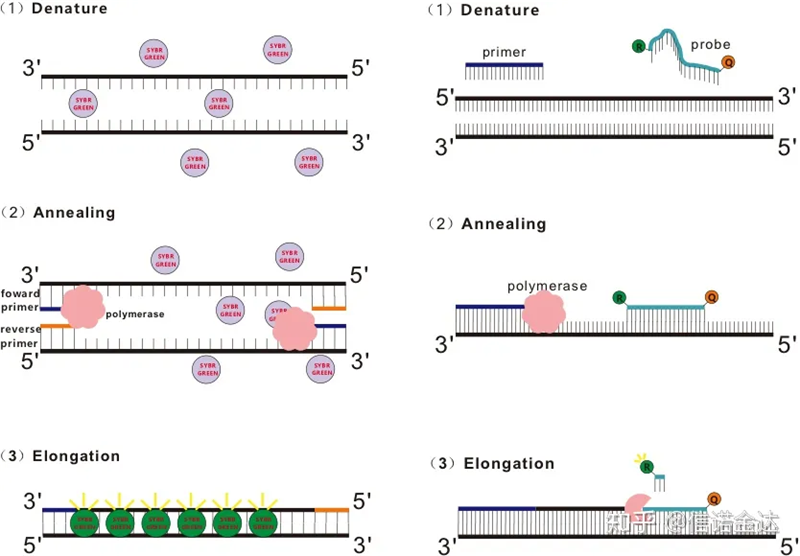
Рис. 1 Принцип qRT-PCR
Дизайн грунтовки
Принципи:
Праймери повинні бути розроблені в консервативній області серії нуклеїнових кислот і мати специфічність.
Найкраще використовувати послідовність кДНК, послідовність мРНК також прийнятна.Якщо ні, з’ясуйте дизайн ділянки cds послідовності ДНК.
Довжина флуоресцентного кількісного продукту становить 80-150 п.н., найдовша — 300 п.н., довжина праймера зазвичай становить 17-25 основ, і різниця між праймерами вище та нижче за течією не повинна бути надто великою.
Вміст G + C становить від 40% до 60%, а 45-55% є найкращим.
Значення TM знаходиться в межах 58-62 градусів.
Намагайтеся уникати праймерних димерів і самодімерів (не з’являйтеся більше 4 пар послідовних комплементарних основ) структура шпильки, якщо це неминуче, зробіть ΔG<4,5 кДж/моль* Якщо ви не можете переконатися, що гДНК було видалено під час зворотної транскрипції Чисто, найкраще спроектувати праймери інтрона *3′ кінець не можна модифікувати, і щоб уникнути багатих областей AT, GC, уникайте T/C, A/G суцільна структура (2-3) праймери та не-
специфічна Гомологія гетерогенно ампліфікованої послідовності переважно становить менше ніж 70% або має гомологію 8 комплементарних основ.
База даних:
CottonFGD пошук за ключовими словами
Дизайн грунтовки:
Дизайн праймера IDT-qPCR
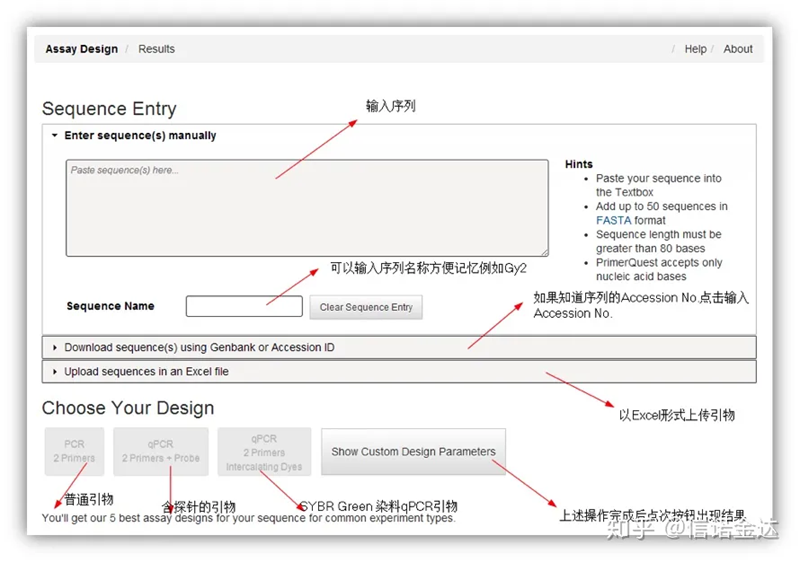
Мал. 2 Сторінка онлайн-інструменту для розробки праймерів IDT
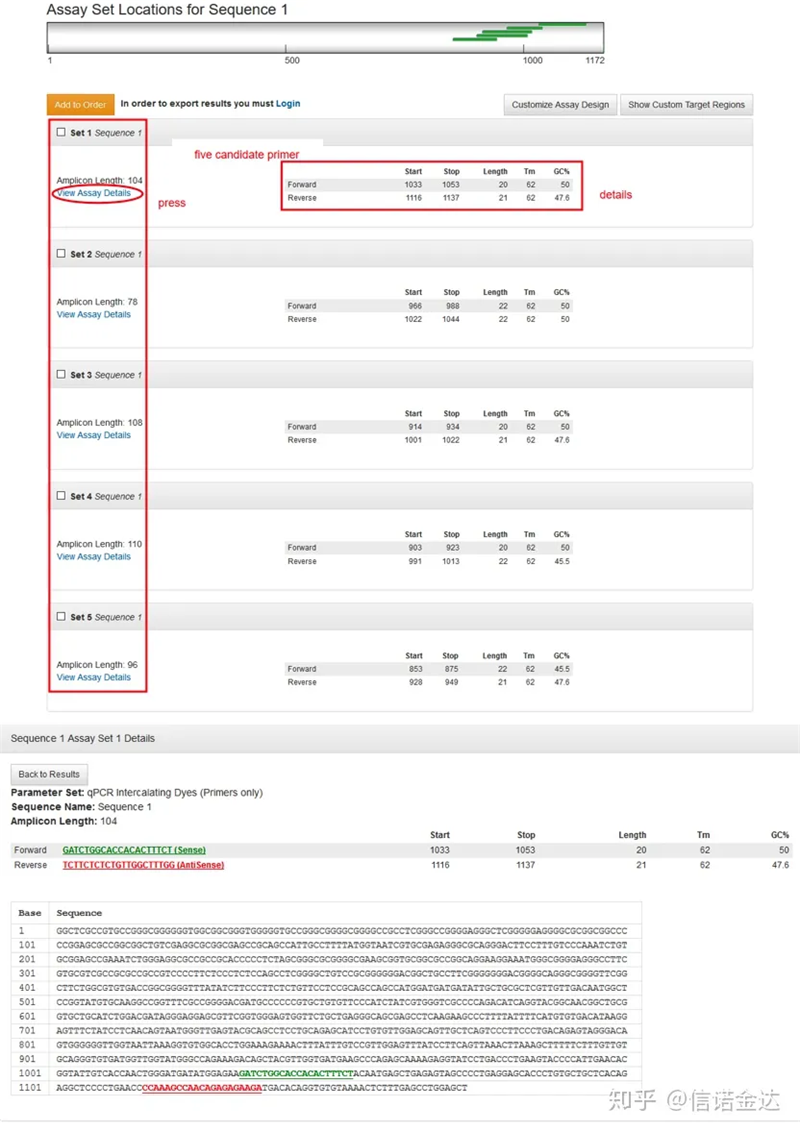
Відображення сторінки результатів Fig3
Дизайн праймерів lncRNA:
lncRNA:ті самі кроки, що і мРНК.
мікроРНК:Принцип методу стовбурової петлі: оскільки всі мікроРНК є короткими послідовностями довжиною близько 23 нт, пряме ПЛР-детектування неможливо виконати, тому використовується інструмент послідовності стовбурової петлі.Послідовність стовбурової петлі — це одноланцюгова ДНК довжиною близько 50 нт, яка сама по собі може утворювати структуру шпильки.3 'Кінець може бути розроблений як послідовність, комплементарна частковому фрагменту мікроРНК, тоді цільова мікроРНК може бути з'єднана з послідовністю стовбурової петлі під час зворотної транскрипції, і загальна довжина може досягати 70 bp, що відповідає довжині ампліфікованого продукту, визначеного за допомогою кПЦР.Дизайн праймера хвостової мікроРНК.
Специфічне виявлення ампліфікації:
Інтернет-база даних вибухів: вибух CottonFGD за подібністю послідовності
Локальний вибух: зверніться до використання Blast+ для локального вибуху, Linux і macos можуть безпосередньо створити локальну базу даних, систему win10 також можна зробити після встановлення ubuntu bash.Створення локальної бази даних вибуху та локального вибуху;відкрити ubuntu bash на win10.
Примітка. Бавовник гористої місцевості та бавовна морських островів є тетраплоїдними культурами, тому результатом вибуху часто буде два або більше збігів.У минулому використання компакт-дисків NAU як бази даних для виконання вибуху, ймовірно, виявило два гомологічні гени лише з кількома відмінностями SNP.Зазвичай два гомологічні гени не можуть бути розділені за допомогою конструкції праймера, тому вони розглядаються як однакові.Якщо є очевидна індель, праймер зазвичай розроблений на інделі, але це може призвести до вторинної структури праймера. Вільна енергія стає вищою, що призводить до зниження ефективності підсилення, але цього не уникнути.
Виявлення вторинної структури праймера:
Кроки:відкрити оліго 7 → ввести послідовність шаблону → закрити підвікно → зберегти → знайти праймер на шаблоні, натиснути ctrl+D, щоб установити довжину праймера → проаналізувати різні вторинні структури, такі як тіло самодімеризації, гетеродимер, шпилька, невідповідність тощо. Останні два зображення на рисунку 4 є результатами тестування праймерів.Результат переднього праймера хороший, немає очевидної димерної та шпилькової структури, немає безперервних комплементарних основ, а абсолютне значення вільної енергії становить менше 4,5, тоді як задній праймер показує безперервність. 6 основ є комплементарними, а вільна енергія становить 8,8;крім того, на 3'-кінці з'являється більш серйозний димер і з'являється димер з 4 послідовних основ.Хоча вільна енергія невисока, 3'-димер Chl може серйозно впливати на специфічність і ефективність ампліфікації.Крім того, необхідно перевірити наявність шпильок, гетеродимерів і невідповідностей.
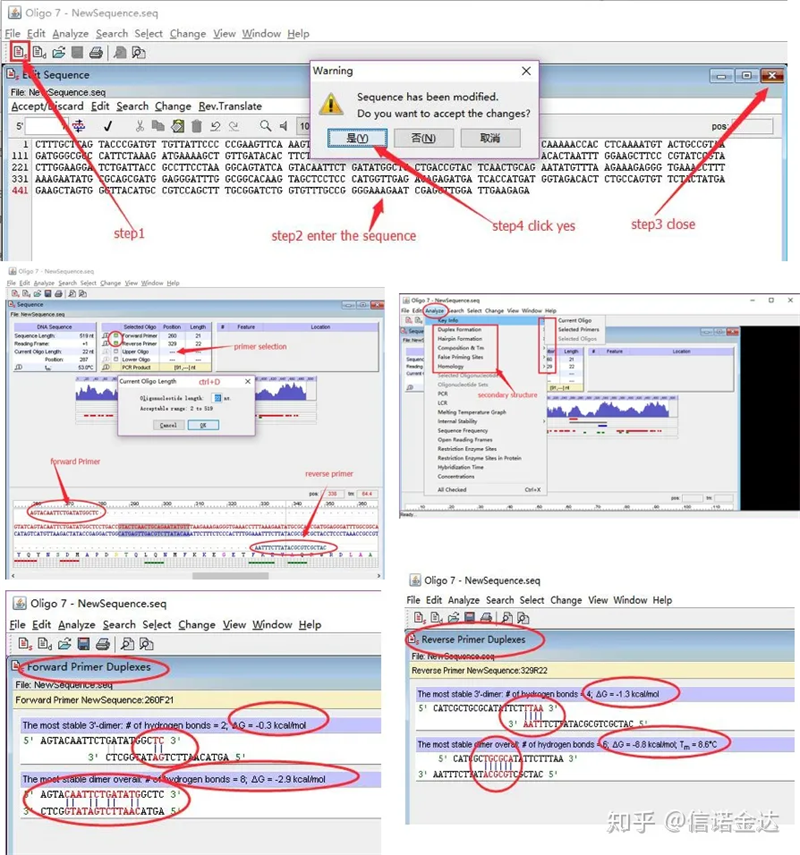
Фіг.3 Результати виявлення оліго7
Виявлення ефективності посилення:
Ефективність ампліфікації реакції ПЛР серйозно впливає на результати ПЛР.Крім того, у QRT-PCR ефективність ампліфікації особливо важлива для кількісних результатів.Видаліть інші речовини, машини та протоколи в реакційному буфері.Якість праймерів також має великий вплив на ефективність ампліфікації qRT-PCR.Щоб забезпечити точність результатів, кількісне визначення відносної флуоресценції та кількісне визначення абсолютної флуоресценції повинні визначати ефективність ампліфікації праймерів.Визнано, що ефективна ефективність ампліфікації qRT-PCR становить від 85% до 115%.Є два методи:
1. Метод стандартної кривої:
a.Змішайте кДНК
b.Градієнтне розведення
c.qPCR
d.Рівняння лінійної регресії для розрахунку ефективності посилення
2. LinRegPCR
LinRegPCR — це програма для аналізу даних RT-PCR у реальному часі, які також називають даними кількісної ПЛР (qPCR) на основі SYBR Green або подібної хімії.Програма використовує дані, не скориговані за базовою лінією, виконує корекцію базової лінії для кожного зразка окремо, визначає вікно лінійності, а потім використовує аналіз лінійної регресії, щоб підігнати пряму лінію через набір даних ПЛР.За нахилом цієї лінії розраховується ефективність ПЛР кожного окремого зразка.Середня ефективність ПЛР на амплікон і значення Ct на зразок використовуються для розрахунку початкової концентрації на зразок, вираженої у довільних одиницях флуоресценції.Введення та виведення даних здійснюється через електронну таблицю Excel.Тільки зразок
потрібне змішування, без градієнту
необхідні кроки:(Візьміть Bole CFX96 як приклад, не зовсім машину з чітким ABI)
експеримент:це стандартний експеримент qPCR.
Вихід даних qPCR:LinRegPCR може розпізнавати дві форми вихідних файлів: RDML або результат кількісної ампліфікації.Фактично, це значення виявлення номера циклу та сигналу флуоресценції машиною в реальному часі, а посилення отримано шляхом аналізу значення зміни флуоресценції ефективності лінійного сегмента.
Вибір даних: Теоретично значення RDML має бути придатним для використання.За оцінками, проблема мого комп’ютера полягає в тому, що програмне забезпечення не може розпізнати RDML, тому я маю вихідне значення excel як вихідні дані.Рекомендується спочатку виконати приблизний відбір даних, наприклад, помилку додавання зразків тощо. Точки можна видалити у вихідних даних (звичайно, ви не можете їх видалити, LinRegPCR проігнорує ці точки на пізнішому етапі)
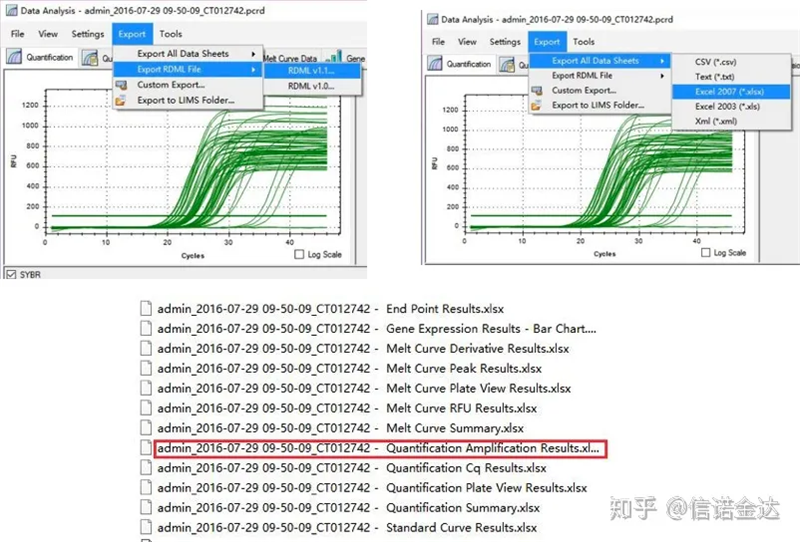
Рис. 5 Експорт даних qPCR
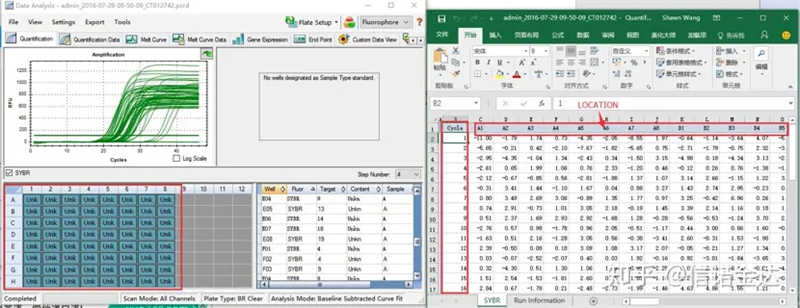
Мал.6 вибір зразків-кандидатів
Введення даних:Відкрийте файл qualification amplification results.xls, → відкрийте LinRegPCR → файл → зчитайте з excel → виберіть параметри, як показано на малюнку 7 → OK → натисніть визначити базові лінії
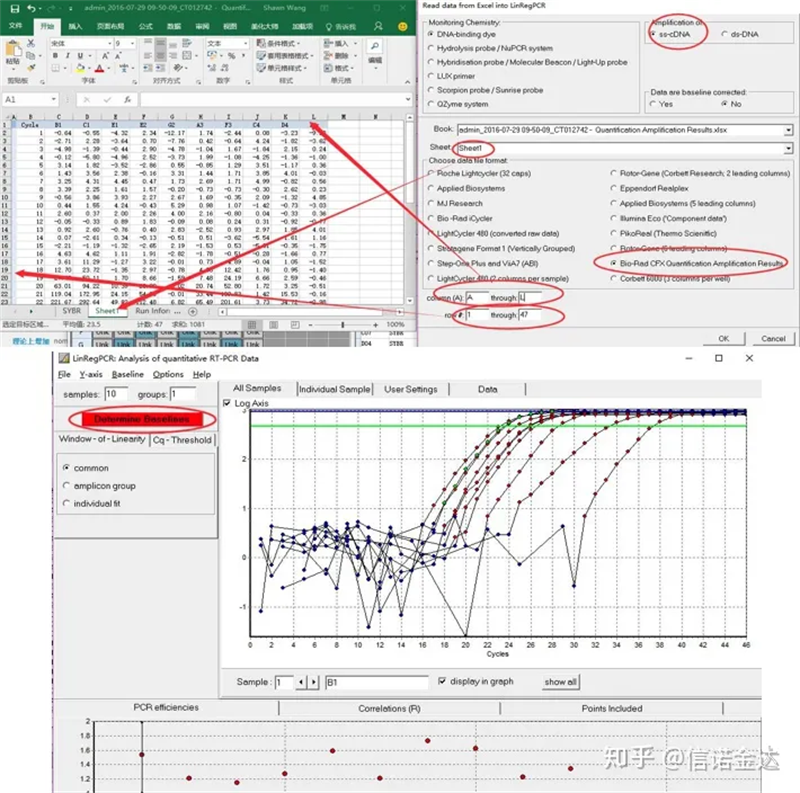
Рис.7 кроки введення даних linRegPCR
Результат:Якщо немає повторення, групування не потрібне.Якщо є повторення, групування можна відредагувати в групуванні зразка, а назву гена ввести в ідентифікатор, після чого той самий ген буде автоматично згруповано.Нарешті клацніть файл, експортуйте файл Excel і перегляньте результати.Буде відображено ефективність ампліфікації та результати R2 для кожної лунки.По-друге, якщо розділити на групи, буде відображено виправлену середню ефективність підсилення.Переконайтеся, що ефективність ампліфікації кожного праймера становить від 85% до 115%.Якщо він занадто великий або занадто малий, це означає, що ефективність підсилення праймера низька.
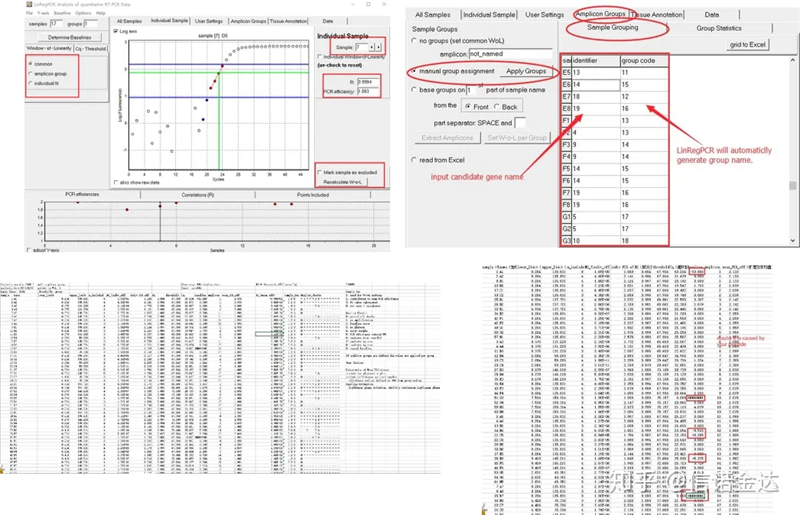
Рис. 8 Результат і вихід даних
Експериментальний процес:
Вимоги до якості РНК:
Чистота:1.72.0 означає, що може бути залишковий ізотіоціанат.Чиста нуклеїнова кислота A260/A230 має бути приблизно 2. Якщо є сильне поглинання при 230 нм, це вказує на наявність органічних сполук, таких як іони фенату.Крім того, його можна виявити за допомогою електрофорезу в 1,5% агарозному гелі.Наведіть маркер, оскільки оцРНК не має денатурації, а логарифм молекулярної маси не має лінійної залежності, і молекулярну масу неможливо правильно виразити.Концентрація: Теоретичноніменше ніж 100 нг/мкл, якщо концентрація занадто низька, чистота, як правило, низька, а не висока
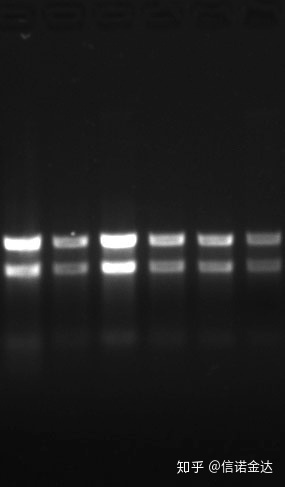
Рис.9 РНК-гель
Крім того, якщо зразок є цінним і концентрація РНК висока, рекомендується аликвотувати його після екстракції та розвести РНК до кінцевої концентрації 100-300 нг/мкл для зворотної транскрипції.впроцес зворотної транскрипції, коли мРНК транскрибується, оліго (dt) праймери, які можуть специфічно зв’язуватися з хвостами polyA, використовуються для зворотної транскрипції, тоді як lncRNA і circRNA використовують випадкові гексамерні (Random 6 mer) праймери для зворотної транскрипції загальної РНК. Для мікроРНК для зворотної транскрипції використовуються специфічні для мікроРНК праймери петлі шиї.Зараз багато компаній випустили спеціальні комплекти для хвостовиків.Для методу стовбурової петлі метод хвоста є більш зручним, високопродуктивним і економить реагенти, але ефект розрізнення мікроРНК однієї родини не повинен бути таким хорошим, як метод стовбурової петлі.Кожен набір для зворотної транскрипції має вимоги до концентрації ген-специфічних праймерів (стеблових петель).Внутрішнє посилання, яке використовується для мікроРНК, — U6.У процесі інверсії стовбурової петлі трубку U6 потрібно перевернути окремо, а передню та задню праймери U6 слід додати безпосередньо.І circRNA, і lncRNA можуть використовувати HKG як внутрішні посилання.ввиявлення кДНК,
якщо немає проблем з РНК, кДНК також має бути в порядку.Однак, якщо ми прагнемо досконалості експерименту, найкраще використовувати внутрішній еталонний ген (Reference gene, RG), який може відрізняти gDNA від cds.Як правило, RG є геном домашнього господарства., HKG), як показано на малюнку 10;У той час я створював соєвий запасний білок і використовував актин7, що містить інтрони, як внутрішню еталонну основу.Розмір ампліфікованого фрагмента цього праймера в гДНК становив 452 п.н., а в разі використання кДНК як матриці — 142 п.н.Тоді результати тесту виявили, що частина кДНК насправді була забруднена gDNA, і це також довело, що немає проблем із результатом зворотної транскрипції, і його можна використовувати як матрицю для ПЛР.Немає сенсу проводити електрофорез у агарозному гелі безпосередньо з кДНК, і це дифузна смуга, що не є переконливим.
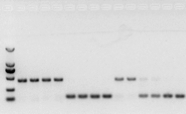
Мал. 10 Виявлення кДНК
Визначення умов КПЦРзагалом немає проблем відповідно до протоколу набору, головним чином на етапі значення tm.Якщо деякі праймери неправильно розроблені під час розробки праймерів, що призводить до великої різниці між значенням tm і теоретичними 60°C, рекомендується кДНК Після змішування зразків провести градієнтну ПЛР з праймерами та намагатися уникати встановлення температури без смуг як значення TM.
Аналіз даних
Звичайний метод кількісної обробки відносної флуоресценції ПЛР в основному відповідає 2-ΔΔCT.Шаблон обробки даних.
Супутні товари:
ПЛР у реальному часіTM –Такман
ПЛР у реальному часіTM –СИБР ГРІН І
RT Easy I (Майстер Премікс для синтезу кДНК першого ланцюга)
RT Easy II (основний премікс для синтезу кДНК першого ланцюга для кПЦР)
Час публікації: 14 березня 2023 р



